Modifying the RMG Databases¶
Editing the Thermodynamic Database¶
As mentioned in Section Primary Thermo Library, it is possible to override the default thermodynamic values and substitute your own thermodynamic data.
Basic Structure of the Thermo Database¶
The thermodynamics database consists of three sections, each of which is an ASCII file that can be edited to alter the information. This description applies to non-radical groups. There are other tree/library/dictionary files for radical groups, ring corrections, and other corrections. The nomenclature is different, but they would be edited in a similar way. The files of interest are located in the directory: $RMG/database/RMG_database/thermo_groups/. The three files are described below.
Dictionary File¶
Group_Dictionary.txt contains the name and adjacency list (structure) for all of the nodes contained within the thermo tree. The nomenclature within all three files must be identical. The asterisk “*” denotes the central atom in the group for which the group value is defined. The format for each line in the adjacency list is: atom #, * if central atom, element, # of radicals (0, 1, 2) on element, bonding:
Cb-(Cd-Cdd-Cd)
1 * Cb 0 {2,S}
2 Cd 0 {1,S} {3,D}
3 Cdd 0 {2,D} {4,D}
4 C 0 {3,D}
Tree File¶
Group_Tree.txt defines the tree structure of the database. The nodes at any particular level are defined by including the text “Lx:” prior to the name of the group at that node, where “x” is a number corresponding to the level in the tree. A sample is given below:
L0: R
L1: C
L2: Cbf
L3: Cbf-CbCbCbf
L2: Cb
L3: Cb-H
L3: Cb-Os
L3: Cb-C
L4: Cb-Cs
L4: Cb-Cd
L5: Cb-(Cd-Od) // Cb-CO
L5: Cb-(Cd-Cd)
L6: Cb-(Cd-Cd) // Cb-Cd
L4: Cb-Cb
L2: Ct
Note that the indentation is not necessary because it is the “Lx:” that the computer reads, but is very helpful in making these files human readable.
Library File¶
Group_Library.txt is the archive of the actual data associated with a given group in the tree and dictionary. There are 15 (space or tab separated) fields in the thermo library to describe the group. The units for enthalpy are kcal/mole, and the units for entropy and heat capacity (Cp) are cal/mole-K. The columns are described in the table “Library File Definitions”.
| Column | What it contains |
|---|---|
| 1 | A unique number; this does not correspond to any other part of the thermo database, but numbering sequentially is most logical |
| 2 | Group name; same as in tree and dictionary |
| 3-4 | Enthalpy and Entropy at 298K |
| 5-11 | Cp at T = 300, 400, 500, 600, 800, 1000, and 1500K |
| 12-13 | dH and dS: absolute uncertainties in the enthalpy/entropy estimates |
| 14 | dCp: absolute uncertainty in the Cp estimate (no temperature consideration) |
| 15 | Comments Section: usually citing the source of the data or comments on the reliability |
A sample entry can be found in Section Editing the Data for an Existing Thermo Functional Group.
Thermo Database and Adjacency List Notation¶
In general, the thermo database uses what are known as function group elements. Function group elements serve to define the atom and its bonding environment. These definitions serve to simplify the adjacency lists of groups and allow for more general descriptions of groups. The notation used in the database is shown below. New functional group elements cannot be added to RMG in a simple way, as they must be hard-coded into the RMG software with the appropriate properties.
If you examine the file Group_Dictionary.txt, you will see that these groups are used extensively, even more so that the actual atoms C, H, or O. You will also see that groups can be defined using a bracketed notation, which simply means that either atom/functional group element will generate this node. For example:
Cb-Cd
1 * Cb 0 {2,S}
2 {Cd,CO} 0 {1,S}
In this example, the second “element” is actually either Cd or CO. The reason that either Cd or CO will generate the same node is because both fall under the more general Cd definition, which is a carbon atom with one double and two single bonds. This can also be useful if there is a secondary effect and all that matters is that there is a π-bond present, but the fact that it is C=O or C=C does not matter. This is not used very much in the thermo database, but occurs much more in the kinetics databases where radical delocalization can play a major role in determining the rate constant of a reaction. A table of the possible functional groups can be seen in the table “Functional Group Elements”.
| Symbol | Definition |
|---|---|
| Cs | Carbon bonded to four single bonds |
| Cd | Carbon bonded to a double bond and two single bonds. (The other end of the double bond is carbon) |
| Cdd | Carbon bonded to two double bonds |
| Ct | Carbon bonded to a triple bond and single bond |
| Cb | Carbon bonded to two benzene bonds and a single bond. (The carbon belongs to only one benzene ring) |
| Cbf | Carbon bonded to three benzene bonds (the carbon belongs to two or three benzene rings) |
| CO | Carbon bonded to a double bond and two single bonds. (The other end of the double bond is oxygen) |
| Os | Oxygen bonded to two single bonds |
| Od | Oxygen bonded to a double bond |
| Oa | Oxygen triplet |
| R | Any atom |
| R!H | Any non-hydrogen atom |
Viewing a Thermo Functional Group in the RMGVE¶
Open the RMG Viewer & Editor (RMGVE) by running the RMGVE 20080101.bat file. If your RMG database is located in the $RMGdatabaseRMG_database folder, the following login screen should appear.
The login name is used to document who made what changes in the RMG libraries. After entering your name, press the “Login” button to reveal the RMGVE.
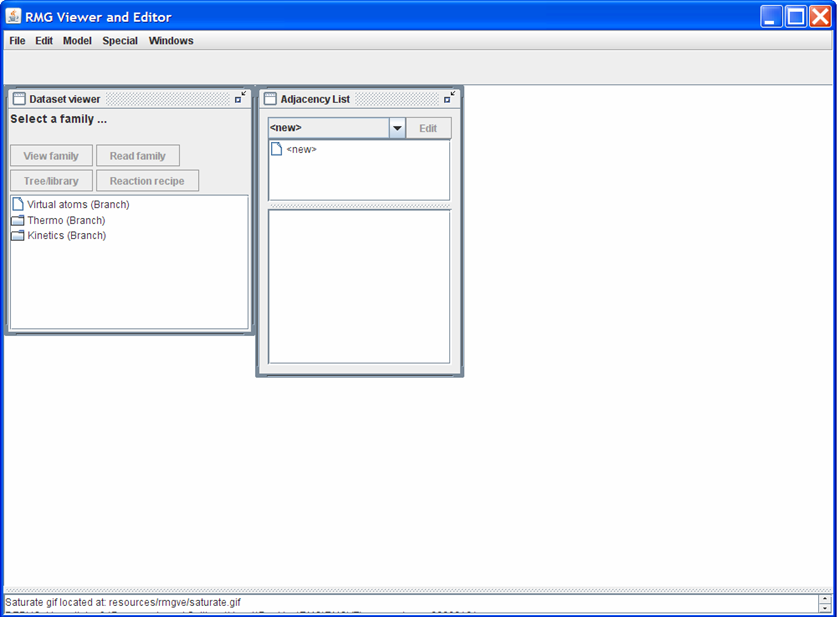
The RMGVE home screen initially has two windows open: the “Dataset viewer” and the “Adjacency List.” In the “Dataset viewer” window, double-click on the “Thermo (Branch)” folder to show the different Thermo Families present in the RMG_Dictionary folder. To load one of the Thermo Families, either:
- double-click on one of the names OR
- highlight one of the names and then push the “Read family” button near the top of the “Dataset viewer” window
Note: Loading a family could take a few minutes, depending on how large the family is.
Once the family is loaded, a new window will pop up. The name of the window corresponds to the name of the Thermo family that was read. In the screen shot shown below, we have chosen to load the “Group” family. Use the scroll bar to see the list of functional groups contained in the “Group” family. To visualize what the different functional groups look like, highlight one of the names and then push the “View” button near the top of the “Group” window. For instance, if we select the functional group name “Cds-OdCsH,” the RMGVE should look as follows.
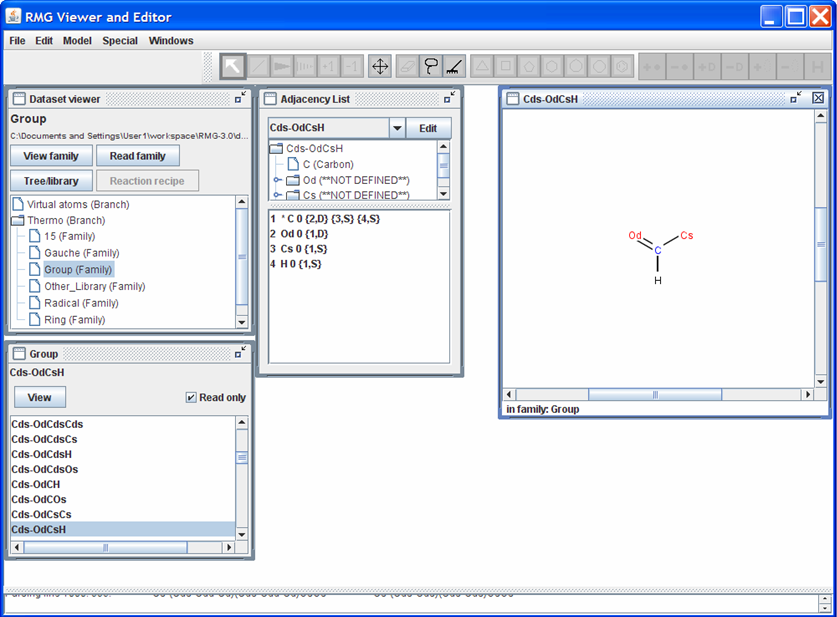
Notice that the “Adjacency List” window is no longer blank but now contains the adjacency list for the functional group “Cds-OdCsH”. Furthermore, a window entitled “Cds-OdCsH” opens which contains a visualization of the functional group.
Note
Notice also that a toolbar appears above the “Dataset viewer,” “Adjacency List,” and “Cds-OdCsH” windows. This toolbar may be used to edit the molecule in the “Cds-OdCsH” window. For instance, click on the icon with the four arrows (pointing up, right, down, and left). Now click on one of the atoms in the “Cds-OdCsH” window and drag it to another location within the window. To restore (clean) the structure of the functional group, click on the icon containing the picture of a rake.
Editing the Data for an Existing Thermo Functional Group¶
A leaf in the thermochemistry tree may be edited using the RMG Viewer & Editor (RMGVE). For instructions on how to add a leaf to a thermochemistry tree, please refer to the section below entitled “Adding Additional Nodes to the Thermo Database”.
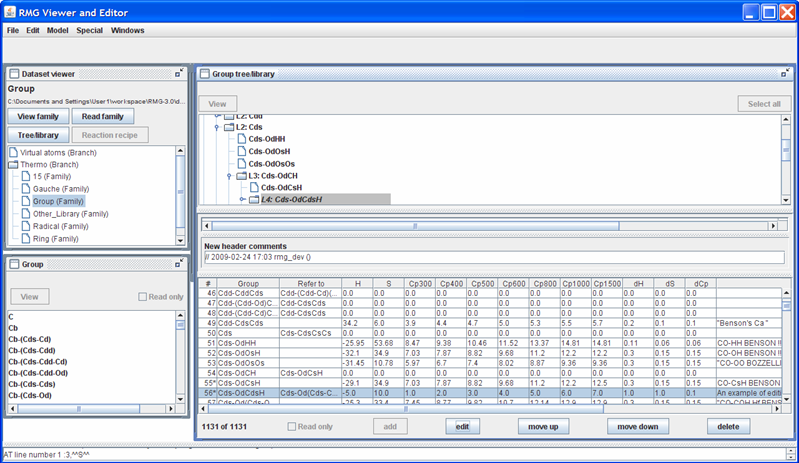
Returning to the “Dataset viewer” window, notice the “Tree/library” button has been enabled. Click on this button to show the data for the highlighted thermo family. After some re-arranging of the windows, the screen should look as follows.
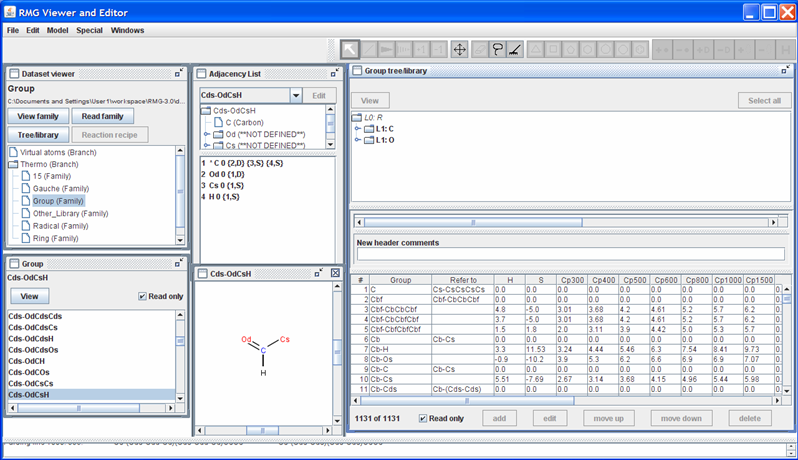
The top half of the new “Group tree/library” window contains the “Group” thermo family tree; the bottom half contains the “Group” thermo family library. Suppose we want to change the thermo value for the “Cds-OdCsH” functional group. To do so, we can navigate the tree in the top half of the “Group tree/library” window by opening (double-clicking) the folders corresponding to the “Cds-OdCsH” functional group:
- L1: C
- L2: Cds
- L3: Cds-OdCH
- Cd-OdCsH
After navigating the tree, the RMGVE should look like the following snapshot.
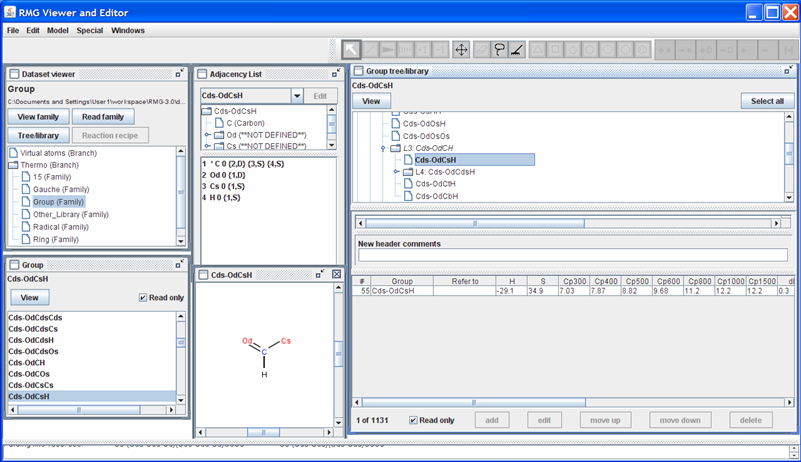
Notice that RMG has thermodynamic data for this functional group. If you scroll to the right (using the scroll bar near the bottom of the “Group tree/library” window) far enough, you will see the “Notes” column. This column is used to document where the data came from. In this case, the thermodynamic data comes from the Benson defined group O=CH-Cs. Furthermore, it has been noted that the Cp1500 was assumed to be the Cp1000 value.
Suppose we had a better estimate for the Cp1500 value (e.g. 12.5 cal/mol/K). To edit this leaf, uncheck the “Read only” field, highlight the row, and push the “Edit” button; the screen should look as follows.
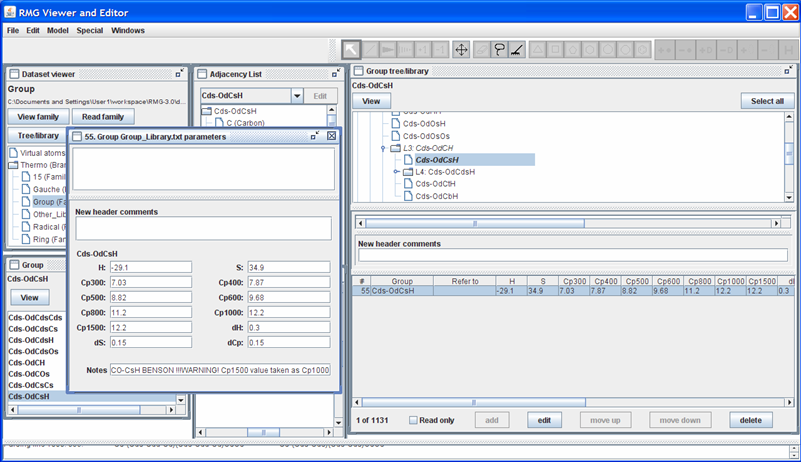
Enter the new thermochemical data (Cp1500 = 12.5) in the appropriate field. The field “Notes” will be added to the thermochemical data for the “Cds-OdCsH” functional group at the end of the line (after the 12 data entries). The field “New header comments” will be placed above the thermochemical data for the “Cds-OdCsH” functional group. Upon changing any of the thermochemical data fields, a line in the “New header comments” will automatically appear, containing your login name and the current date and time. The box immediately above the “New header comments” field shows how the RMGVE will edit the current data in the Group_Dictionary.txt file. Once all data has been entered, close the window.
Returning to the “Group tree/library” window, notice the highlighted row. The thermochemical data entries have been updated and a star (*) has been placed next to the “55” value in the first column. The star (*) reflects that a change has been made.
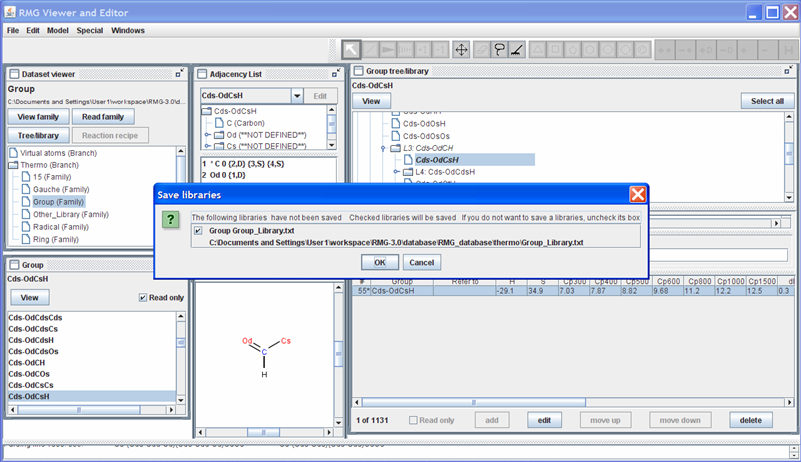
When you have completed making changes to the thermochemistry database, close the RMGVE. Upon doing so, you will be asked which changes you would like to accept and save to the RMG_database folder. Before pushing the “OK” button, ensure that all of the changes you want to be saved have a checkmark next to them. You will receive a message stating whether the edits were saved successfully or not. Pushing “OK” in this window will close the RMGVE.
Note
Notice the RMGVE directs you to the location of the file that is about to be changed.
Viewing the changed thermochemical data¶
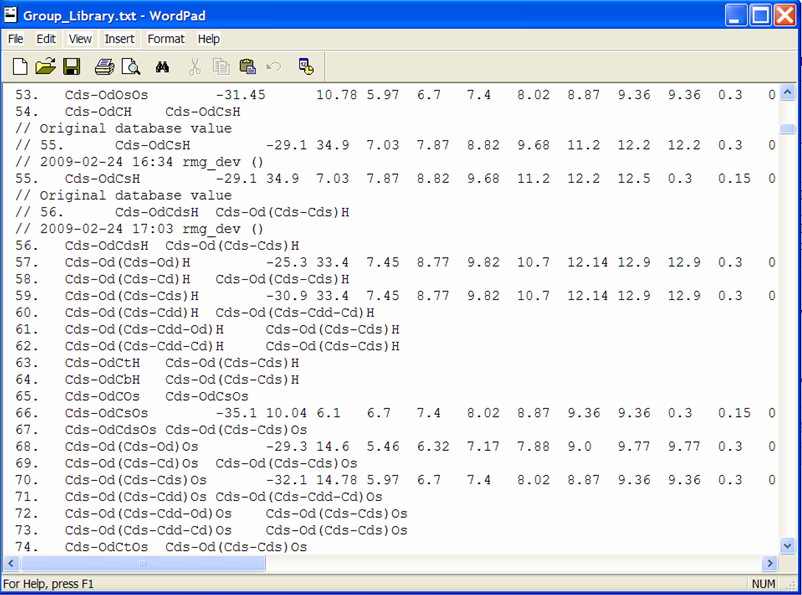
To view the thermochemical data that was just edited, open the file from the previous screenshot: $RMG/database/RMG_database/thermo_groups/Group_Library.txt. Scrolling down to the entry #55, we see the old and new thermochemical data for the functional group “Cds-OdCsH”.
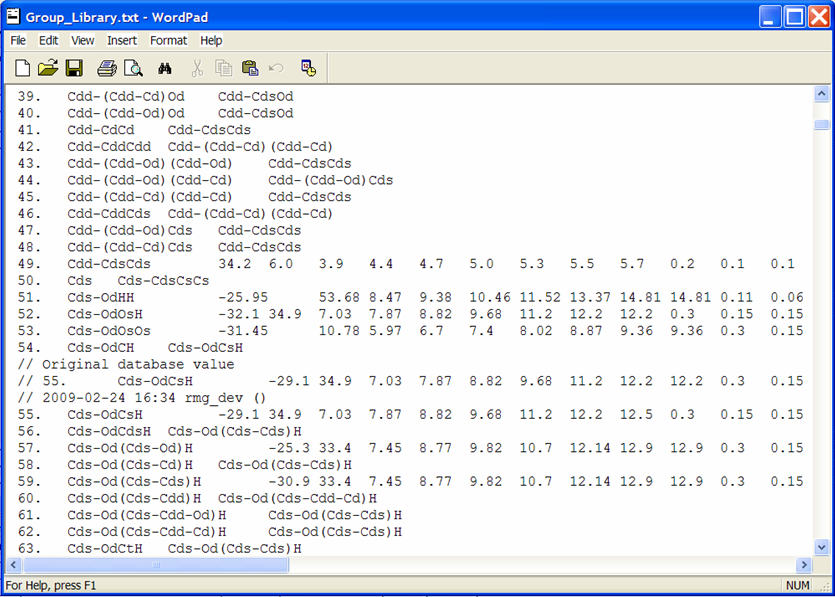
Rather than explicitly entering data for each leaf, one may also refer one functional group to another, if you believe they should be the same. This is done by putting the name of the group that has the same thermo parameters in column 3 and leaving columns 4-15 blank. An example of this is entry #56 where we see that this leaf does not contain thermodynamic data but rather points to the functional group “Cds-Od(Cds-Cds)H,” entry #59. Effectively, if RMG encounters the group and finds the name of another group instead of numerical values, it will assign the current group the same values that occur in the referred group:
56. Cds-OdCdsH Cds-Od(Cds-Cds)H
59. Cds-Od(Cds-Cds)H -30.9 33.4 7.45 8.77 etc.
The above example would assume that the group values for #56 are identical to those of #59.
This referencing does not need to be done if the group to which you want to refer lies directly above the current group in the tree, because if the tree does not have a certain node defined it will look back up the tree to find the nearest relative and use those values:
253 Cb-(Os-(Os-Cs)) -2.5 -8.5 etc...
254 Cb-(Os-(Os-H)) Cb-(Os-(Os-Cs))
255 Cb-(Os-(Os-(Cs-OsHH))) Cb-(Os-(Os-Cs))
In this case, the referring of #255 back to #253 is unnecessary because RMG would refer back to #253 by default if it did not find any values for #255. This redundancy will not create any problems within RMG, but it is simply unnecessary.
Warning
The RMGVE can only edit leafs which already have thermochemical data stored for them. In particular:
- The RMGVE does not allow a user to change the “Refer to” field. This change must be performed manually.
- The Group_Library.txt file will not recognize changes made in the RMGVE to leafs that “Refer to” another species. For example, had we entered thermochemical data for entry #56, the RMGVE will show the updates to the H, S, and Cp values in the “Group tree/library” window but will also still show the “Refer to” functional group. After closing the RMGVE and confirming the change to the database, if one opened the Group_Library.txt file and looked at entry #56, you will notice the leaf’s thermodynamic values were not updated.
Please be aware that version control may be a significant issue with the databases and should be addressed early to ensure consistency within the group.
Adding Additional Nodes to the Thermo Database¶
This task is more complicated than the previous example because it involves altering all three database files in a consistent manner, without the help of a Graphical User Interface. It is worth noting that the order in which species are added to the dictionary (_Dictionary.txt file) and library (_Library.txt file) does not matter; however, the position where the new group is added to the tree (_Tree.txt file) is of the utmost importance. It is useful to create the tree and dictionary with items in the same order. This ordering will facilitate cross checking and debugging if needed. Since all files must use the same nomenclature, the ability to search may make consistent ordering unnecessary. The procedure is described below.
Find the appropriate location to place the new group and ensure that the nomenclature is unambiguous and unique. Placing the group in the incorrect location could cause incorrect estimates to be made when the tree is being searched. Using the RMGVE to navigate the tree structure is a useful way to find the location for your new group.
Add the line of text to the Group_Tree.txt file in the following form, where “x” is the appropriate level. The following example is for a O-O-H off of a benzene ring.:
Lx: Cb-(Os-(Os-H))
Using the same name as in the tree, append the file Group_Dictionary.txt to define the structure of the group and its atom center (denoted by the asterisk):
Cb-(Os-(Os-H)) 1* Cb 0 {2,S} 2 O 0 {1,S} {3,S} 3 O 0 {2,S} {4,S} 4 H 0 {3,S}Add the new group to the Group_Library.txt file using the same nomenclature and whatever thermo data you have for the group. The format was shown in the previous section.:
2374 Cb-(Os-(Os-H)) 3.5 10.0 ... "I added this b/c ..."
Editing the Kinetics Database¶
RMG’s kinetics database may also be viewed and edited using the RMGVE. From the RMGVE’s home screen, double-click on the “Kinetics (Branch)” folder in the “Dataset viewer” window and then double-click on the “kinetics (Family)” folder. Scroll down to the family entitled “H_Abstraction.” Highlight this family and push the “Read family” button. All 4 buttons in the “Dataset viewer” window should now be active.
- “Read family”: This button reads in the functional groups pertaining to the highlighted library and displays them in a new window; the name of the new window corresponds to the thermo/kinetic family name. (Option available for both thermo and kinetics).
- “Tree/library”: This button opens a window that displays the thermochemical data for the highlighted library. (Option available for both thermo and kinetics).
- “Reaction recipe”: This button opens a window that displays the definition of the reaction family. (Option available ONLY for kinetics).
- “View family”:
Viewing a Reaction Recipe in the RMGVE¶
Push the “Reaction recipe” button. Your screen should look like the following snapshot (after rearranging and resizing the windows)
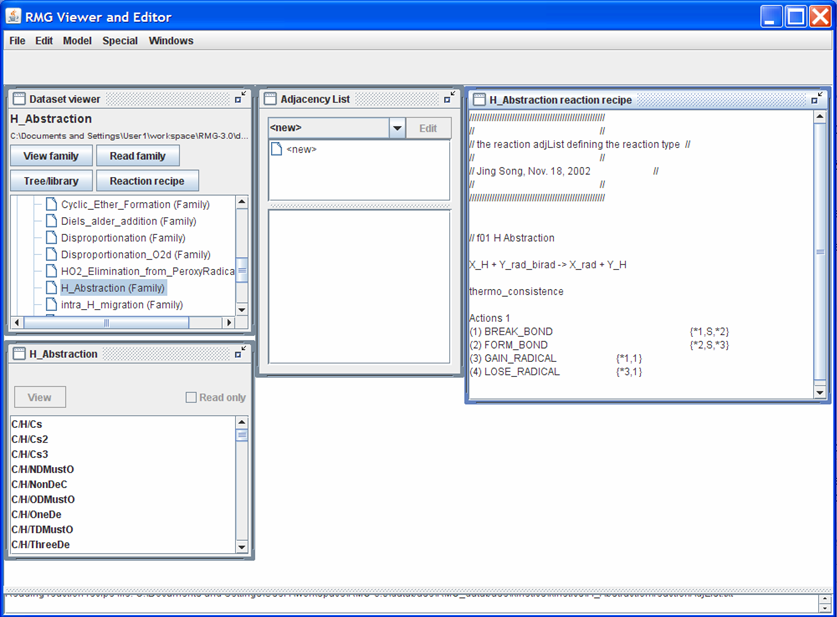
Looking at the “H_Abstraction reaction recipe” window: The first non-commented line shows the generic reaction: X_H + Y_rad_birad -> X_rad + Y_H. The next uncommented line informs RMG how to handle the reverse reaction. Some common occurences you will find include:
- “thermo_consistence”: RMG will use the equilibrium constant to compute the reverse reaction rate
- “none”: RMG assumes the reaction is irreversible
- “(f_): <rxnFamilyName>”: RMG may use the forward reaction rates stored in the <rxnFamilyName> folder
The next set of uncommented lines describes the changes in the reacting molecules’, “X_H” and “Y_rad_birad”, connectivity graphs to form the products, “X_rad” and “Y_H”. For the “H_Abstraction” reaction family, a single bond (“S”) is broken between atoms “1” and “2” and a single bond (“S”) is formed between atoms “2” and “3”. Furthermore, atom “1” gains a radical while atom “3” loses a radical.
For a better understanding of what the atom numbering means, click on one of the species in the “H_Abstraction” window. If you click on the species “C/H/Cs3”, your screen should look like the following snapshot
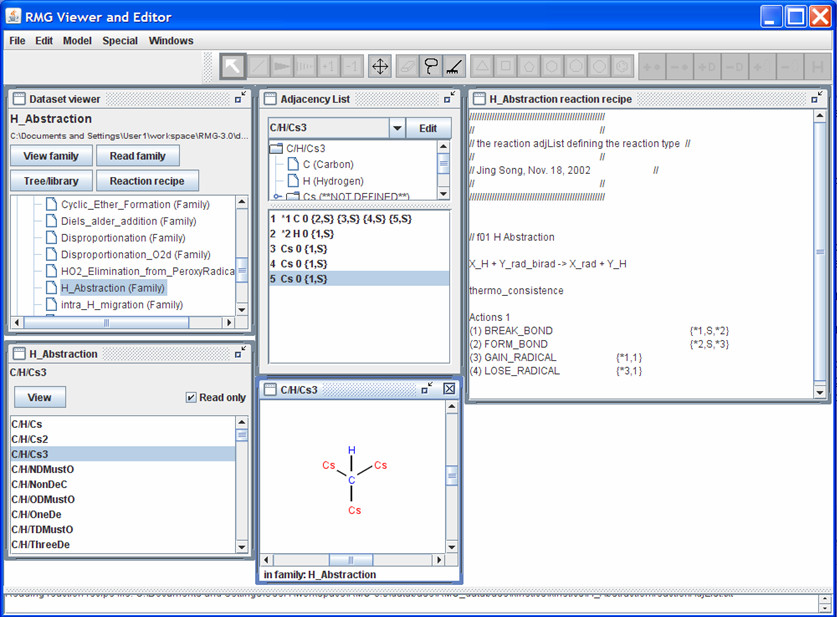
Looking at “C/H/Cs3” and “Adjacency List” windows reveals the identity of atoms “1” and “2”. Looking at the “Adjacency List” window, notice the atoms that have the #* notation between the counting index and the elemental symbol. The number before the * corresponds to the number in the reaction recipe. The atoms of interest are also shown in blue in the “C/H/Cs3” window.
Editing a Reaction Family using the RMGVE¶
Returning to the “Dataset viewer” window, click the “Tree/library” button. The bottom-half of this window contains the following kinetic information:
Groups: The columns entitled “Group 1”, “Group 2”, etc. represent the structure of the reactant(s).
Temp: This column contains the Temperature range (in units of Kelvin) over which the kinetic data is valid.
A, n, a, E0: These columns contain the modified Arrhenius parameters (A, n, E0) and the Evans-Polanyi coefficient (a). “A” has units of “mol,cm3,s” and “E” has units of “kcal/mol”.:
The Evans-Polanyi principle states that for a series of closely-related reactions, a linear relationship between the activation energy (Ea) and the enthalpy of reaction (delHr) is sometimes observed and can be expressed as: Ea = C1 + a*delHr where C1 and a are empirically-derived constants. For RMG, we assume C1 = 0.dA, dn, da, dE0: These columns contain the error for the Arrhenius parameters and Evans-Polanyi coefficient. If the number is reported with a star (*) in front of it, this represents a multiplicative error; else, the error is additive.
Rank: This column contains the confidence the RMG developers have in the kinetic data on a scale of 1 to 5, with 1 being “very confident” and 5 being “rough estimate”.
Notes: This column contains notes on where the kinetic data came from. The reaction library is not as well-documented as the thermo library.
Suppose we want to add a reaction rate to RMG for the following reaction:
H3C-CH2-CH3 + *CH3 = H3C-*CH-CH3 + CH4
The first step would be to identify which of RMG’s reaction templates the reaction of interest corresponds to. For our case, the reaction template we want is “H_Abstraction”. The next step is to find the pair of nodes in the tree that correspond to the reaction of interest. Looking at the tree, we need to identify a “X_H” and “Y_rad_birad”. For the example, “X” is the central carbon of propane and “Y” is the methyl radical. Traversing down the “X_H” tree leads us to the following node:
L1: X_H
L2: Cs_H
L3: C_sec
C/H2/NonDeC
If you are ever uncertain of the structure of one of the nodes, highlight it and push the “View” button to open a window with a 2-d graphical depiction of the species. Traversing the “Y_rad_birad” tree leads us to:
L1: Y_rad_birad
L2: Y_rad
L3: Cs_rad
C_methyl
Your screen should look something like the following
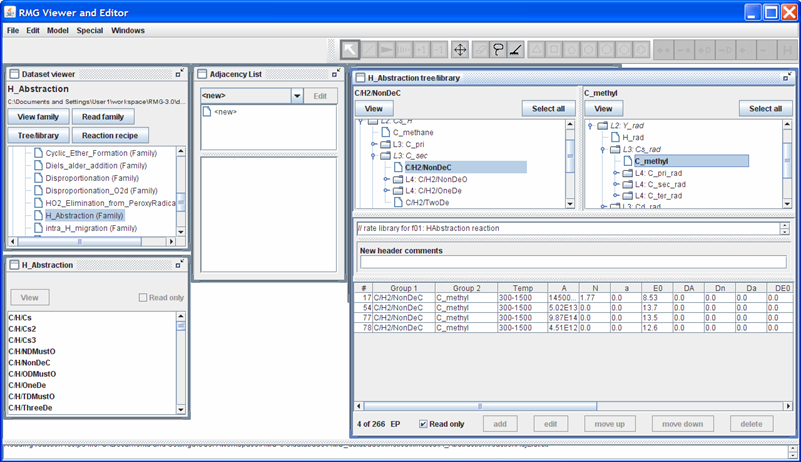
Notice that RMG contains 4 different sets of kinetic data for this reaction. There are two explanation for this:
The reaction rates all pertain to the same reaction: The different values could pertain to experimental vs. theoretical values or differing basis sets in the theoretical calculations (e.g. using an ab initio quantum chemistry sofware package)
The reaction rates pertain to different reactions: Notice that the “X_H” leaf is only defined as “C/H2/NonDeC”. Thus, one of the reaction rates could correspond to the methyl radical abstracting a Hydrogen atom from:
- propane to form the iso-propyl radical
- butane to form the sec-butyl radical
- ...
For RMG’s reaction library, the order in which the reactions appear in the rateLibrary.txt file is significant. RMG will use the kinetic data for the first instance of the reaction of interest that it finds. If you click on one of the 4 reaction rates, you’ll notice that the RMGVE gives you the option of “edit”ing or “delete”ing the entry (as it did for the Thermo library) but it also gives you the option to “move up” or “move down” the reaction rate.
We can also add a new entry, by pushing the “add” button. For example, let’s suppose we have the following modified Arrhenius parameters for the reaction mentioned above: A = 1.27E14 cm3/mol/s, n = 1.3, E = 10.9 kcal/mol over the temperature range 300-1500K. Push the “add” button, then push the “edit” button, and then fill in the popup window with the kinetic data. After closing the popup window, your screen should look like this
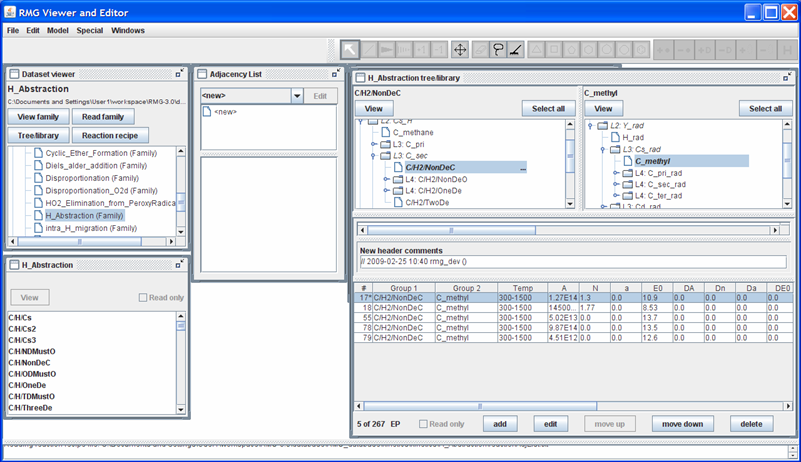
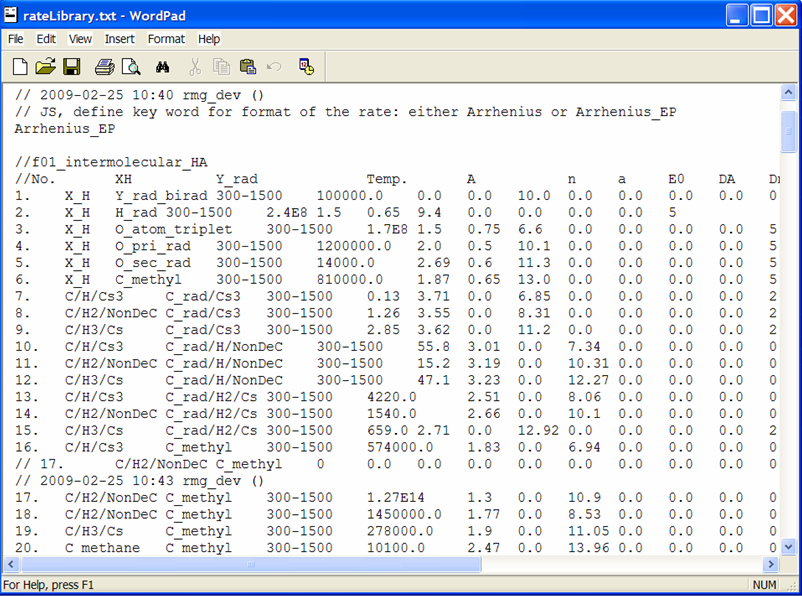
Close the RMGVE. A popup window will ask if you want to save the changes you have made. If you leave the box checked and then open the $RMG/database/RMG_database/kinetics_groups/H_Abstraction/rateLibrary.txt file, you should see that the file has been updated.
Creating a Primary Reaction Library / Seed Mechanism¶
Primary reaction libraries and seed mechanisms are all stored in $RMG/databases/RMG_Database/kinetics_libraries/
A primary reaction library / seed mechanism may consist of five files:
- species.txt
- reactions.txt
- 3rdBodyReactions.txt
- lindemannReactions.txt
- troeReactions.txt
The species.txt file is required. All other files are optional, and if present, must include the Unit declaration and Reaction headings.
The species.txt file is a series of molecular names and connectivity lists, analogous to the formats used in the input file condition.txt. All species present in the four remaining files must be given a structure in species.txt. The structure should have the same format as the adjacency list shown in Creating a condition file. Please note that the names that are used in the reaction library will be those used throughout the mechanism. In this manner, the user can adopt a preferred nomenclature for individual species.
The reactions.txt file defines the standard non pressure-dependent reaction. The file has the structure shown in the following sample. Comments are denoted with “//” and are ignored by the RMG parser.:
// Define the units // Units allowed for A are: "mol/liter/s" or "mol/cm3/s" // Units allowed for E are: "kcal/mol", "cal/mol", "kJ/mol", or "J/mol" Unit: A: mol/cm3/s E: kcal/mol Reactions: // The format is: // R1 + R2 + R3 <=> P1 + P2 + P3 A n E dA dn dE // where R1, R2, R3, P1, P2, P3 are species; A, n, and E are the Arrhenius // parameters, and dA, dn, dE are the errors in those parameter (normally // additive, but can also be multiplicative if a * comes before the number). // A "<=>" or "=" represents a reversible reaction and a // "=>" or "->" represents an irreversible reaction. O2 + CO = CO2 + O 1.26E13 0.00 196.90 *1.7 0 0
The 3rdBodyReaction.txt file lists reactions involving a third body (bath gas). A sample file, with a 3rd-body reaction, is listed below. The first line defines the reaction using the same format as in reactions.txt. The next line lists collision efficiencies for various bath-gas species that scale the concentrations of particular species when calculating the total bath gas concentration. In the example below, CH4 is particularly effective as a 3rd body, and its effective concentration is tripled.:
// Define the units // Units allowed for A are: "mol/liter/s" or "mol/cm3/s" // Units allowed for E are: "kcal/mol", "cal/mol", "kJ/mol", or "J/mol" Unit: A: mol/cm3/s E: kJ/mol Reactions: CO + O + M = CO2 + M 1.54E15 0.00 12.56 *1.2 0 0 N2/0.4/ O2/0.4/ CO/0.75/ CO2/1.5/ H2O/6.5/ CH4/3.0/ C2H6/3.0/ AR/0.35/ // the first line defines the reaction and Arrhenius parameters, // while the second gives the scaling factors for different bath gas species // which contribute to [M].
The lindemannReaction.txt file allows specifying pressure-dependent reactions, according to the Lindemann expression. The format of each reaction is the same as is used by CHEMKIN. For example:
Unit: A: mol/cm3/s E: cal/mol Reactions: O + CO (+M) <=> CO2 (+M) 1.800E+10 .000 2385.00 0.0 0.0 0.0 H2/2.00/ O2/6.00/ H2O/6.00/ CH4/2.00/ CO/1.50/ CO2/3.50/ C2H6/3.00/ Ar/ .50/ LOW/ 6.020E+14 .000 3000.00/ // the first two lines are similar to the 3rdBodyReaction.txt format // the next line specifies the low pressure limit Arrhenius parameters
The troeReactions.txt file allows specifying pressure-dependent reactions according to the Troe expression. The format of each reaction is the same as is used by CHEMKIN. For example:
Unit: A: mol/cm3/s E: kJ/mol Reactions: C2H2 + H (+M) = C2H3 (+M) 8.43E12 0.00 10.81 *1.2 0 0 N2/0.4/ O2/0.4/ CO/0.75/ CO2/1.5/ H2O/6.5/ CH4/3.0/ C2H6/3.0/ AR/0.35/ LOW / 3.43E18 0.0 6.15 / TROE / 1 1 1 1231 / // the first three lines are similar to the lindemannReaction.txt format // the next line specifies the 3 or 4 Troe parameters // in the order: a, T***, T*, T** (the last parameter is optional).
![]()
Documentation Contents
- Modifying the RMG Databases
- Editing the Thermodynamic Database
- Editing the Kinetics Database
- Creating a Primary Reaction Library / Seed Mechanism
Previous topic
Next topic
Estimating Species Thermochemistry